ExtractCgMLST - Determine the set of loci that constitute the core genome
Requirements to define a core genome MLST (cgMLST) schema
cgMLST schemas are defined as the set of loci that are present in all strains under analysis or, due to sequencing/assembly limitations, >95% of strains analyzed. In order to have a robust definition of a cgMLST schema for a given bacterial species, a set of representative strains of the diversity of a given species should be selected. Furthermore, since cgMLST schema definition is based on pre-defined thresholds, only when a sufficient number of strains have been analyzed can the cgMLST schema be considered stable. This number will always depend on the population structure and diversity of the species in question, with non-recombinant monomorphic species possibly requiring a smaller number of strais to define cgMLST schemas than panmictic highly recombinogenic species that are prone to have large numbers of accessory genes and mobile genetic elements. It is also important to refer that the same strategy described here can be used to defined lineage specific schemas for more detailed analysis within a given bacterial lineage. Also, by definition, all the loci that are not considered core genome, can be classified as being part of an accessory genome MLST (agMLST) schema.
Determine the loci that constitute the cgMLST
Determine the set of loci that constitute the core genome based on loci presence thresholds.
Basic Usage
chewBBACA.py ExtractCgMLST -i /path/to/AlleleCallResultsFolder/results_alleles.tsv -o /path/to/OutputFolder
Parameters
-i, --input-file (Required) Path to the TSV file that contains the allelic profiles determined
by the AlleleCall module.
-o, --output-directory (Required) Path to the directory where the process will store the output
files.
--t, --threshold (Optional) Genes that constitute the core genome must be in a proportion
of genomes that is at least equal to this value. Users can provide multiple
values to compute the core genome for multiple threshold values (default:
[0.95, 0.99, 1]).
--s, --step (Optional) The allele calling results are processed iteratively to evaluate
the impact of adding subsets of the results in computing the core genome. The
step value controls the number of profiles added in each iteration until all
profiles are included (default: 1).
--r, --genes2remove (Optional) Path to a file with a list of gene IDs to exclude from the
analysis (one gene identifier per line) (default: False).
--g, --genomes2remove (Optional) Path to a file with a list of genome IDs to exclude from
the analysis (one genome identifier per line) (default: False).
Outputs
The output folder contains the following files:
Presence_Abscence.tsv- allele presence and absence matrix (1 or 0, respectively) for all the loci found in the-ifile (excluding the loci and genomes that were flagged to be excluded).
mdata_stats.tsv- total number and percentage of loci missing from each genome.
cgMLST<threshold>.tsv- a file for each specified threshold that contains the matrix with the allelic profiles for the cgMLST (already excluding the list of loci and list of genomes passed to the--rand--gparameters, respectively).
cgMLSTschema<threshold>.txt- a file for each specified threshold that contains the list of loci that constitute the cgMLST schema. This file can be passed to the--glparameter of the AlleleCall module to perform allele calling only for the loci in the list.
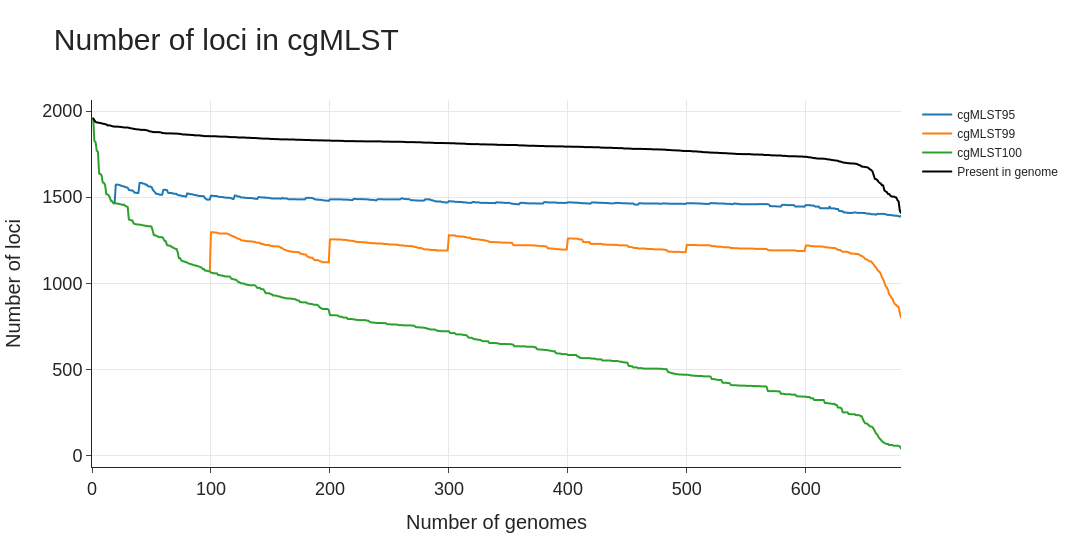
cgMLST.html- HTML file with a line plot for the number of loci in the cgMLST per threshold. Also includes a black line with the number of loci present in each genome that is added to the analysis.
Important
The ExtractCgMLST module masks the allelic profiles, which removes all INF- prefixes and substitutes non-EXC and non-INF classifications by 0.
Example of the plot created by the ExtractCgMLST module based on the allelic profiles for 680 Streptococcus agalactiae genomes:
Note
The matrix with allelic profiles created by the ExtractCgMLST process can be imported into PHYLOViZ to visualize and explore typing results.
Workflow of the ExtractCgMLST module
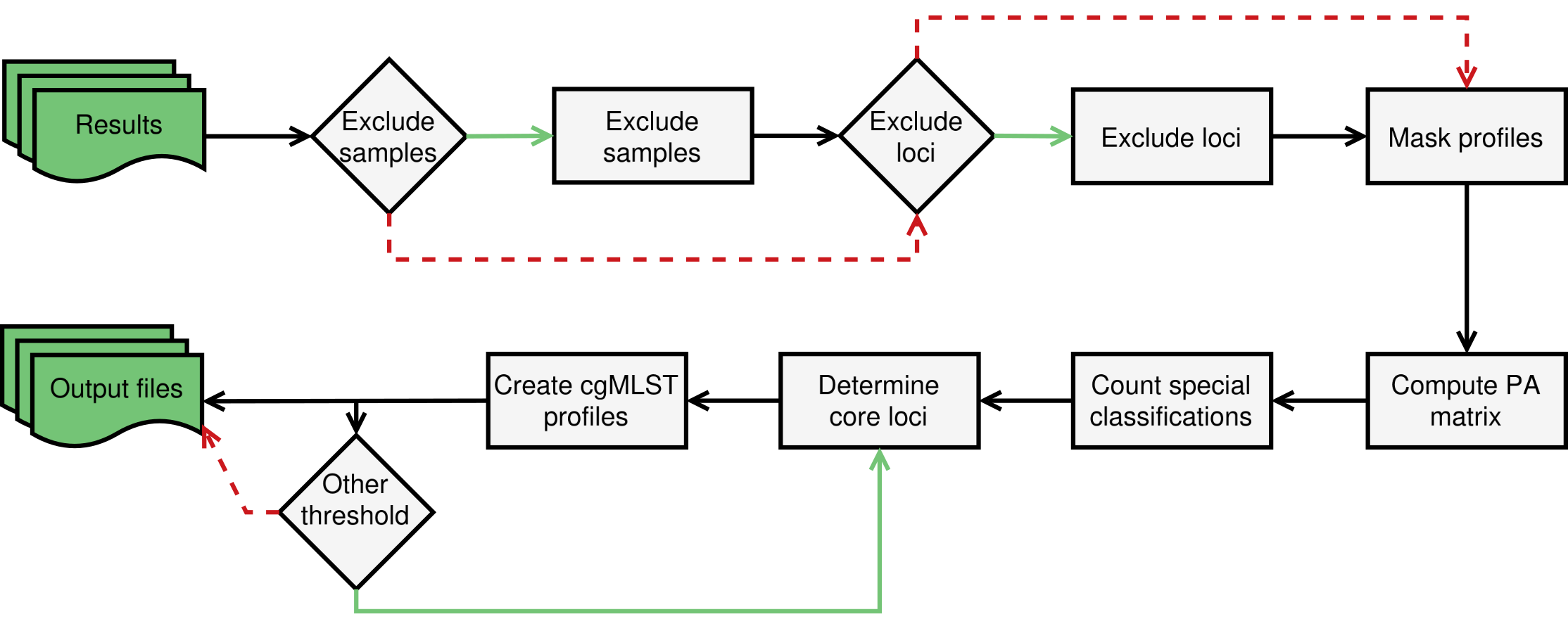
The ExtractCgMLST module determines the set of core loci based on the allelic profiles determined by the AlleleCall module. Brief description of the workflow:
The process starts by excluding loci and samples from the analysis based on lists of loci and samples provided by the user. This allows users to filter out low-quality samples and problematic loci that would affect the determination of the core genome.
The filtered allelic profiles are masked to remove the INF- prefixes from newly inferred alleles and substitute special classifications by
0.The masked profiles are used to compute a loci presence-absence matrix and count the number of special classifications per sample.
The presence-absence matrix is also used to determine the set of core loci based on the default loci presence thresholds of 0.9, 0.95 and 1, or based on threshold values specified by the user.
The process creates output files with the list of loci and allelic profiles per threshold and creates an HTML file with a scatter plot representing the core genome size variation for each threshold.